张洪彬教授课题组完成了Spirotryprostatin A、Spirotryprostatin B及6-Methoxyspirotryprostatin B的高效不对称合成。在该合成工作中他们建立了有机催化的二羰基吡嗪(DKP)骨架的合成新方法,利用该方法合成了一批多官能团取代的含二羰基吡嗪化合物;该方法简单高效,提供了DKP化合物的类型,有利于进一步的官能团化。关键化合物的结构经过NOE实验及X-单晶衍射实验确定(图1)。
利用建立的有机催化的二羰基吡嗪骨架合成方法,对Spirotryprostatins及Fumitremorgins生物碱的合成路线进行研究,成功利用该方法完成了Spirotryprostatin类型螺环天然产物的全合成,形式合成了Fumitremorgins结构的天然产物(图2)。6-Methoxyspirotryprostatin B为首次合成,仅8步反应(从已知化合物计算为6步反应),18%的总收率。Spirotryprostatin B的合成为9步反应(从已知化合物计算为7步反应),19%的总收率,spirotryprostatin A为9步反应,17%的总收率。该路线是目前为止最高的总收率。文献中合成该类物质最短路线为6步,但只有4.6%的总收率,最长路线25步,3.4%总收率。该合成路线同时是目前立体选择性最佳的路线。该论文目前已在《欧洲化学》发表(Chem. Eur. J. 2019, 25, 3005)。
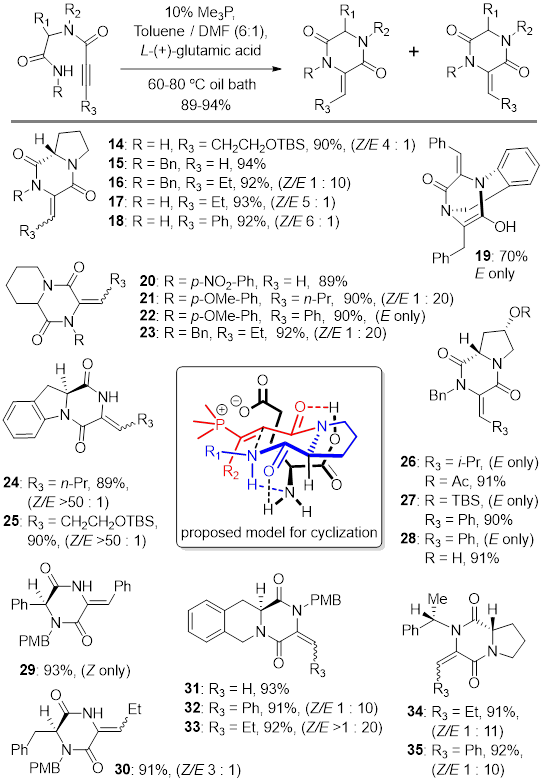
图1 二羰基吡嗪(DKP)骨架的合成新方法
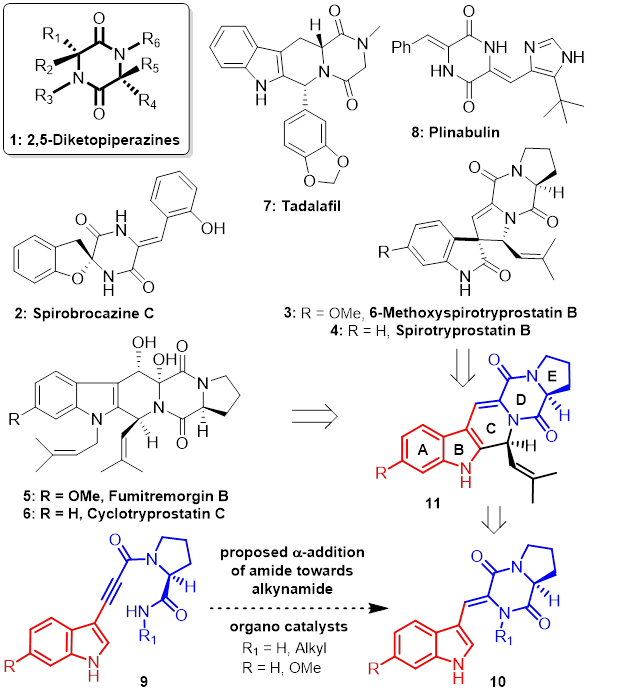
图2 Spirotryprostatins及Fumitremorgins生物碱的合成研究
在天然产物合成方面, 张洪彬教授课题组还完成了六氢吡咯吲哚生物碱(+)-Nocardioazine B的全合成(图3)。该部分工作已经在美国化学会的刊物《有机化学》上发表(J. Org. Chem. 2018, 83, 14507)。
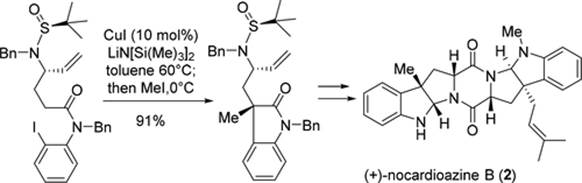
图3 六氢吡咯吲哚生物碱(+)-Nocardioazine B的全合成
羊晓东/张洪彬教授团队与宾夕法尼亚大学Patrick J. Walsh教授课题组合作,在自由基环化串联反应合成苯并呋喃的研究领域取得重要进展。该成果以“Synthesis of Benzofuran Derivatives through Cascade Radical Cyclization/ Intermolecular Coupling of 2‐Azaallyls”(2019, 58, 2826-2830,DOI: 10.1002/anie.201812369)为题发表在国际化学领域权威期刊《德国应用化学》(Angewandte Chemie International Edition)。该论文首次报道了串联自由基环化/分子间偶联反应合成苯并呋喃衍生物的新方法。
苯并呋喃结构单元作为重要的生物活性基团广泛存在于天然产物分子和药物分子中,在抗肿瘤、抗病毒、抗菌、抗炎、抗氧化以及对心血管系统的保护作用等方面表现出良好的生理活性。因此,其相关合成及构效关系研究一直是医药、农药及化学工业领域的研究热点之一。仅2015年,就34种FAD批准的临床药物是苯并呋喃衍生物,包括达芬那新,维拉佐酮和雷美替胺等。苯并呋喃基乙胺是苯并呋喃中一类重要的衍生物,是α-肾上腺素受体拮抗剂的合成前体,其引起了合成工作者的兴趣。然而,前人报道的合成路线冗长、产率低、高温、使用过渡金属催化剂/配体/危险试剂(钯、镍、重氮甲烷、氢化铝锂等),且涉及自由基环化串联反应非常少。因此,发展一种简洁、高效、安全的方法用于合成苯并呋喃基乙胺是有必要的。
该方法以原料易得的2-氮杂烯丙基化合物和碘代芳基丙二烯基醚为反应物,在双(三甲基硅基)氨基钠为碱、乙二醇二甲醚(DME)为溶剂的反应体系中,室温条件下,首次实现了自由基环化串联分子间偶联反应,应用于苯并呋喃乙胺类化合物的合成(图4)。该方法具有良好的普适性,同时可以拓展合成一系列常规合成方法难以制备的多功能化、多(杂)环苯并呋喃衍生物,也可以扩环合成一系列苯并六元或七元氧杂环衍生物。值得一提的是,该串联反应不需要过渡金属催化、无配体,避免了在后处理过程中过渡金属杂质分离的难度,同时降低反应成本,避免了来自可持续性的挑战问题,这将有助于该工作今后在制药领域中的应用。
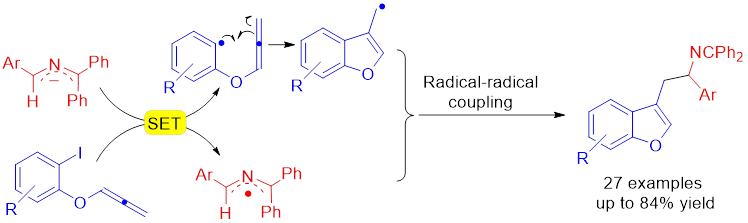
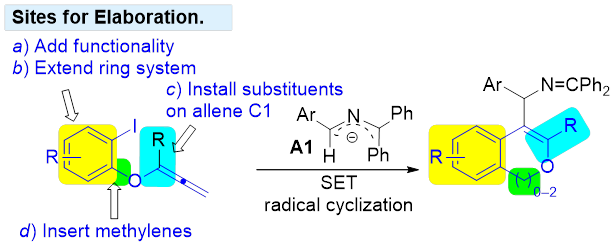
图4 自由基环化串联分子间偶联反应合成苯并呋喃
羊晓东/张洪彬教授团队通过过渡金属催化的C(sp3)—C(sp2)偶联反应对吡啶甲基胺/醚类化合物,实现了分子间的烯基化、芳基化等官能团化新方法(见图1.4)。1) 发展了一种钯催化下吡啶甲基胺与烯基卤化物的烯基化C(sp3)—C(sp2)偶联反应 (33个例子,收率可达98%);2) 发展了一种镍催化下吡啶甲基醚的化学选择性芳基化和串联芳基化/[1,2]-Wittig重排C(sp3)—C(sp2)偶联反应 (55个例子,收率可达96%)。该系列反应实现了目标化合物的克级制备,可应用于后续药物分子及天然产物的合成研究。文章分别发表在《Adv. Synth. Catal.》及《Org. Chem. Front.》上。同时利用二氯甲烷为试剂,发展的吖啶的不对称合成也是一个很有用处的中间体,可应用于天然产物及类天然杂环化合物的合成(图5)。
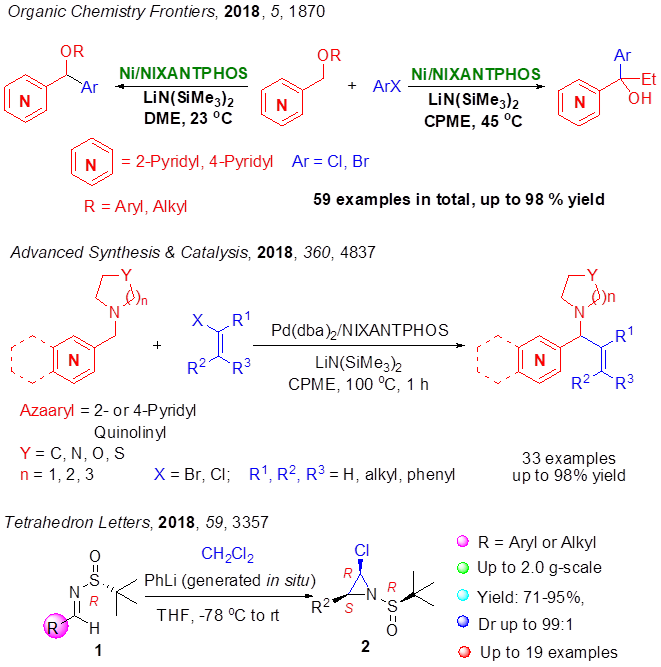
图5 类天然杂环化合物的合成方法
夏成峰课题组对二甲酰胺类溶剂进行了研究,发现在氟代磺酰胺的存在下,醋酸亚铜可以将胺基邻位的甲基上的氢原子通过氧化的方式经自由基生成碳正离子,与芳胺反应生成胺缩醛,并进一步与腈基反应生成a-胺基腈(图6)。a-胺基腈是很多药物和天然产物中的重要基团,它们也可以轻易地转化成其它的药物活性基团。虽然著名的斯特雷克反应也能合成a-胺基腈,但合成的化合物类型却非常局限。该方法具有更广的底物普适性,可一步生成不同结构的a-胺基腈。研究成果发表在《Chem. Commun. 2018, 54, 2854》上。
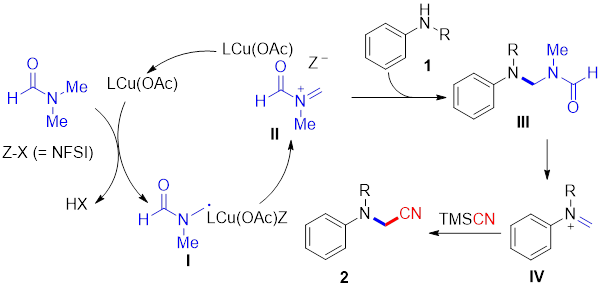
图6 a-胺基腈的合成
夏成峰课题组同时发现该铜催化的反应体系对醚类溶剂也同样具有很好的活化作用。以四氢呋喃溶剂为研究对象,醋酸亚铜可以将醚中与氧相邻的碳原子上的氢原子通过氧化的方式经自由基生成碳正离子,并与芳基胺反应生成缩酮结构。当体系中有腈类试剂存在时,会进一步催化缩酮开环,生成链状的a-胺基腈。在该研究中,首次发现环状的醚可以转化成链状分子,并且反应具有优良的底物兼容性,进一步拓展了该反应在药物分子合成中的价值(图7)。研究成果发表在《Chem. Commun. 2018, 54, 11033》。
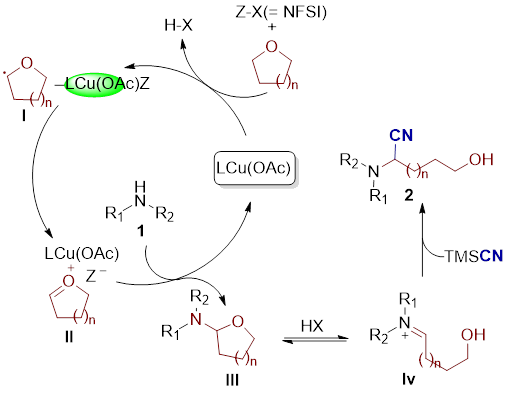
图7 铜催化醚中惰性的碳氢键活化
